前言導讀
肌萎縮性脊髓側索硬化症(ALS),也就是我們俗稱的「漸凍症」,是一種極其殘酷的疾病。在科學界,研究漸凍症一直有個巨大的障礙:我們缺乏一個理想的「動物替身」。
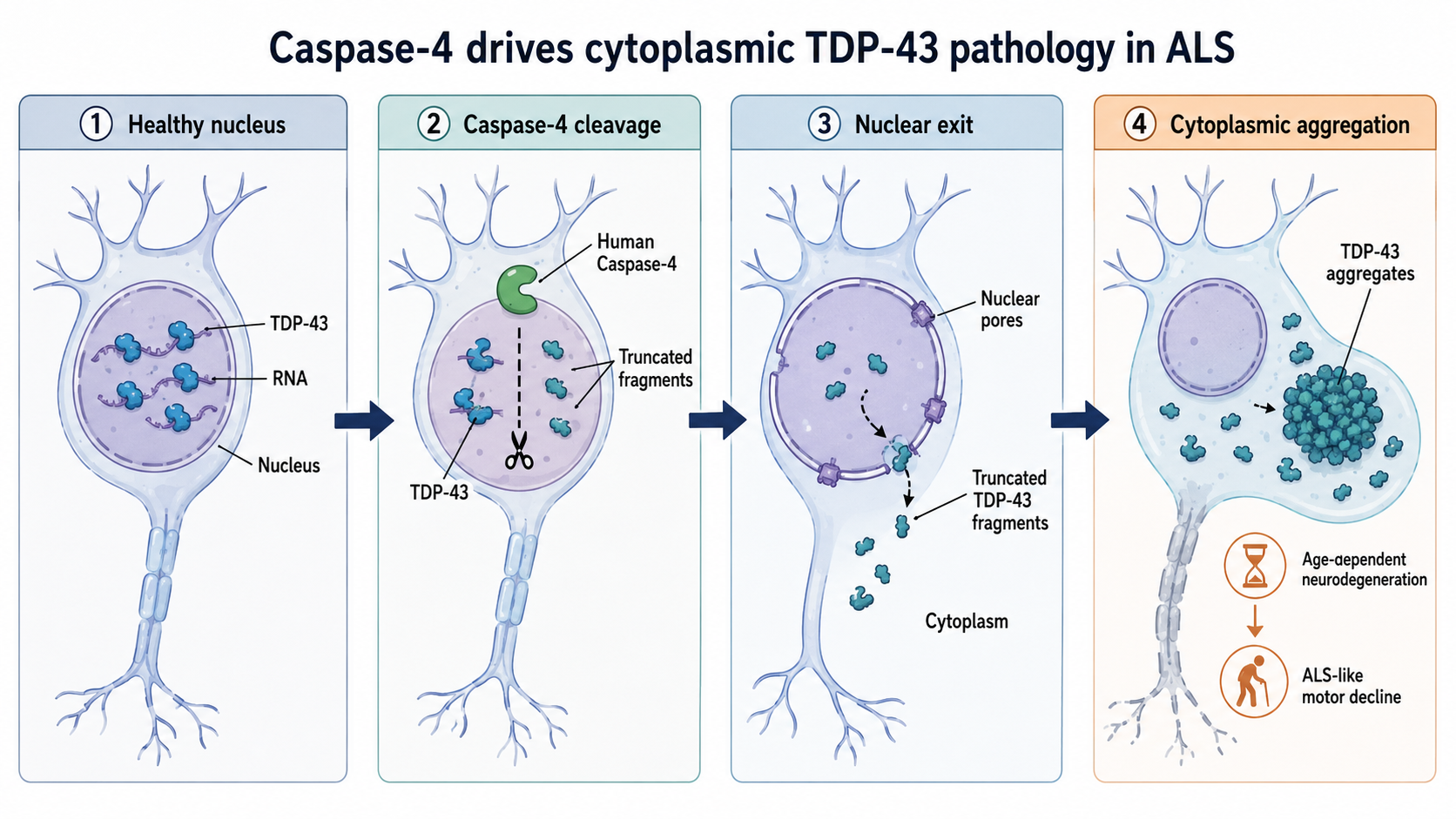
在超過 90% 的漸凍症患者體內,科學家發現一種名為 TDP-43 的蛋白質發生了異常。正常情況下,TDP-43 應該待在細胞核(細胞的指揮中心)裡工作,但在患者的神經細胞中,它卻會「離家出走」,跑到細胞質(細胞核外的空間)堆積成一團團的垃圾。這不僅讓指揮中心癱瘓,堆積物還具有毒性。然而,過去科學家在實驗小鼠身上,很難重現這種「蛋白質位置放錯」的關鍵現象,導致研發新藥時往往在動物身上有效,到了人類臨床試驗卻失敗。
這篇研究的重要性在於,研究團隊找到了一個關鍵的「壞人」——Caspase-4 (CASP4)。這是一種靈長類(如人類、猴子)才有的酵素,而一般老鼠並沒有。這項研究的核心直覺非常簡單:既然老鼠沒有這個壞人,所以牠們的 TDP-43 不會亂跑,那如果我們把這個人類的壞人基因放進老鼠體內呢?這個嘗試成功開啟了漸凍症研究的新篇章。
完整故事
這場科學探索的起點,源於一個長期困擾醫學界的難題:為什麼我們無法在實驗室裡完美模擬漸凍症?雖然科學家早已鎖定 TDP-43 是致病的主角,但傳統的基因轉殖小鼠模型,要嘛病徵不夠像,要嘛就是無法呈現出 TDP-43 從細胞核移到位在細胞質堆積的典型病理特徵。
研究團隊開始尋找線索。他們注意到人類與老鼠在演化上的一個細微差異:人類擁有一種稱為 Caspase-4 的酵素,而老鼠卻沒有。在之前的研究中,團隊發現這個酵素就像一把利剪,會把 TDP-43 蛋白質剪斷。被剪斷後的 TDP-43 碎片變得很不穩定,會直接從細胞核「偷溜」出去,並在細胞質中糾結成塊。
為了驗證這個想法,研究人員決定打造一個全新的實驗武器:Caspase-4 轉基因小鼠。他們透過基因工程,讓小鼠也表現出這種人類才有的酵素。結果令人振奮且驚訝:隨著年齡增長,這些小鼠開始出現明顯的運動功能障礙,牠們的肌肉變得無力,動作變得遲緩,這與漸凍症患者的症狀高度相似。
當研究人員進一步解剖觀察小鼠的神經細胞時,他們發現了尋找已久的「冒煙槍」(關鍵證據):這些小鼠神經元中的 TDP-43 真的發生了移位,它們離開了細胞核,在細胞質中形成了大量的堆積物。更重要的是,這些小鼠體內的基因表現特徵,與人類的「散發性漸凍症」(sporadic ALS)患者驚人地一致。
這項發現不僅僅是創造了一個新的模型,更指出了治療的新方向。研究團隊嘗試使用一種名為反義寡核苷酸(ASO, Antisense Oligonucleotide)的新型藥物療法。這種藥物就像是精準的導彈,專門攔截並抑制 Caspase-4 的產生。實驗結果顯示,接受治療的小鼠,其 TDP-43 的異常堆積明顯減少,神經毒性也得到緩解。
這對科學與醫療界意義重大。我們現在不僅擁有了一個更像人類患者的「動物替身」來測試新藥,還證實了 Caspase-4 是導致漸凍症病理變化的重要推手。這為占絕大多數的散發性漸凍症患者,帶來了開發精準治療藥物的全新希望。
Guided Introduction
Amyotrophic Lateral Sclerosis (ALS), often called Lou Gehrig’s disease, is a devastating condition that causes the death of neurons controlling voluntary muscles. For decades, one of the biggest hurdles in ALS research has been a lack of "realistic" animal models. In over 90% of human ALS cases, a protein called TDP-43—which is supposed to stay inside the cell’s nucleus to manage genetic information—malfunctions. It leaks out into the cytoplasm (the outer area of the cell) and forms toxic clumps.
The core problem is that standard laboratory mice do not naturally show this "leaking and clumping" behavior. When scientists try to study ALS in these mice, the results often don't translate to humans because the underlying biology is different. This paper introduces a breakthrough based on a clever intuition: humans have a specific "molecular scissor" called Caspase-4 that mice lack.
By understanding that this human-specific enzyme is what actually cuts TDP-43 and forces it to leak out of the nucleus, researchers have finally created a mouse model that mimics the actual pathology seen in human patients. This "on-ramp" to understanding the study is simple: to study a human disease in an animal, you first have to give the animal the specific human tools that cause the damage. This research provides a new, more accurate "test subject" for developing the life-saving drugs that ALS patients desperately need.
The Full Story
The story begins with a mystery in the landscape of neurodegenerative disease. While scientists knew that the protein TDP-43 was the primary "bad actor" in most ALS cases, they couldn't figure out why it was leaving its post in the cell nucleus to form dangerous aggregates in the cytoplasm. Because laboratory mice don't naturally develop these specific protein clumps, researchers were essentially trying to solve a puzzle with missing pieces.
The researchers looked for clues in the evolutionary differences between humans and rodents. They focused on Caspase-4, an enzyme found in primates but absent in mice. In previous work, the team discovered that Caspase-4 acts like a set of specialized shears that can chop TDP-43 into smaller fragments. These truncated pieces are unstable; they cannot stay in the nucleus and instead migrate into the cytoplasm, where they stick together and poison the cell.
To test if this was the "missing link" for ALS, the scientists created a new type of transgenic mouse that expresses the human Caspase-4 gene. As these mice aged, the results were striking. Unlike previous models, these mice began to exhibit the classic signs of ALS. Their TDP-43 proteins moved to the cytoplasm and formed inclusions, and the mice developed motor dysfunctions, such as muscle weakness and coordination loss, that grew worse over time.
Furthermore, when the team analyzed the gene expression in these mice, they found it was remarkably similar to the patterns seen in humans with sporadic ALS—the most common form of the disease that occurs without a clear family history. This confirmed that the Caspase-4 mice were not just "sick," but were sick in the exact same way that humans are.
The final chapter of this study offers a glimmer of hope for future medicine. The researchers used a modern therapeutic tool called an antisense oligonucleotide (ASO)—a designer molecule that can "silence" specific genes. By using an ASO to turn off the Caspase-4 enzyme in the mice, they were able to reduce the TDP-43 clumping and protect the neurons from further damage. This suggests that Caspase-4 is not just a marker of the disease, but a viable target for treatment. For the scientific community, this means a better platform for drug testing; for patients, it identifies a new path toward stopping the progression of "the freezing disease."
這是一份根據來源文件整理,針對一般讀者編寫的研究摘要:
1. 一句話總結
這項研究開發出一種新型轉基因小鼠模型,透過表現人類的 Caspase-4 酵素,成功模擬了漸凍症(ALS)患者體內關鍵致病蛋白質 TDP-43 的異常堆積與運動功能障礙。
2. 簡單內容概述
- 研究目的:目前的漸凍症小鼠模型通常無法表現出患者身上常見的「TDP-43 蛋白質從細胞核移到位在細胞質堆積」的現象,這阻礙了對病理機轉與藥物的研究。
- 實驗方法:研究人員開發了一種表現人類 Caspase-4 (CASP4) 基因的轉基因小鼠,並觀察其隨著年齡增長產生的變化。
- 主要發現:
- 這些小鼠的 TDP-43 確實產生了細胞質堆積,且會隨年齡增長出現運動功能障礙。
- 小鼠的基因表現和神經病理特徵,與人類「散發性漸凍症」(sporadic ALS)患者高度相似。
- 使用藥物(反義寡核苷酸,ASO)抑制 Caspase-4,可以改善小鼠的病理症狀並減少神經毒性。
3. 機制邏輯(一步一步說明)
核心致病流程如下:
- 酵素作用:存在於靈長類(及本研究轉基因小鼠)體內的 Caspase-4 酵素被活化。
- 切割蛋白質:Caspase-4 會切割原本待在細胞核內的 TDP-43 蛋白質。
- 位置出錯:被切割後的 TDP-43 碎片無法待在細胞核,而是跑到了細胞質。
- 異常堆積:在細胞質中的 TDP-43 碎片形成堆積物(inclusions),產生神經毒性。
- 導致發病:這最終導致運動神經元的損害,引發如肌肉無力、運動困難等漸凍症症狀。
4. 為什麼重要 / 應用
這項研究的重要性在於它提供了一個更貼近人類病理的動物模型。這讓科學家能更精準地研究「散發性漸凍症」(約佔漸凍症病例的 90% 以上)的發病機制,並用來測試如 ASO 藥物等潛在的治療方案,加速新藥開發。
5. 需要記住的關鍵名詞
- TDP-43 (TAR DNA-binding protein):原本負責處理細胞內遺傳訊息的蛋白質,在漸凍症患者體內會出錯並在細胞質堆積,是主要的致病指標。
- Caspase-4 (CASP4):一種靈長類特有的酵素,本研究發現它是導致 TDP-43 被切割並移位的關鍵推手。
- 漸凍症 (ALS, 肌萎縮性脊髓側索硬化症):一種運動神經元退化導致肌肉萎縮、無力的疾病。
- 反義寡核苷酸 (ASO, Antisense Oligonucleotide):一種新型的基因療法藥物,能精準干擾特定基因的表現(如本研究中用來抑制 Caspase-4)。
1. One-Sentence Summary
This research developed a new transgenic mouse model expressing the human Caspase-4 enzyme to successfully replicate the protein clumping and motor decline characteristic of human ALS.
2. Overview
- Research Goal: To create an animal model that accurately mirrors how the TDP-43 protein malfunctions in human ALS patients, something previous mouse models struggled to do.
- What They Did: Scientists engineered mice to produce human Caspase-4 (CASP4), an enzyme primates have but mice naturally lack, and observed how it affected their nervous systems over time.
- Main Findings:
- The mice developed toxic protein clumps (inclusions) in the cytoplasm of their nerve cells, just like human patients.
- As the mice aged, they experienced progressive motor dysfunction and muscle weakness.
- The genetic changes in these mice closely matched those found in humans with sporadic ALS (the most common form of the disease).
- Using a specialized gene-silencing drug (ASO) significantly reduced the protein damage and protected the nerve cells.
3. Mechanism Logic
The core process of the disease in this model follows these steps:
- Enzyme Activation: The human Caspase-4 enzyme becomes active within the motor neurons.
- Protein Cutting: Caspase-4 acts like molecular scissors, cutting the TDP-43 protein into smaller, broken fragments.
- Leakage: These broken fragments lose their ability to stay inside the cell's "command center" (the nucleus) and leak out into the surrounding area (the cytoplasm).
- Clumping: In the cytoplasm, these fragments stick together to form toxic "trash piles" called aggregates or inclusions.
- Nerve Damage: These clumps interfere with cell health, eventually causing the motor neurons to die and leading to muscle paralysis.
4. Why It Matters / Applications
- Better Drug Testing: Since this mouse model behaves much more like a human ALS patient than previous models, it provides a more reliable platform for testing new medications before they go to human trials.
- Identifying a New Culprit: By proving that Caspase-4 is a "trigger" for the disease, researchers have a clear target for new therapies.
- Validation of ASO Therapy: The study shows that Antisense Oligonucleotides (ASOs) can successfully block this toxic process, offering hope for a potent treatment for sporadic ALS.
5. Key Terms to Remember
- TDP-43: A vital protein that normally stays in the cell nucleus but becomes a primary driver of ALS when it clumps in the cytoplasm.
- Caspase-4 (CASP4): A human enzyme identified in this study as the "scissors" that cut TDP-43 and start the disease process.
- ALS (Amyotrophic Lateral Sclerosis): A progressive neurodegenerative disease that destroys the nerve cells responsible for muscle movement.
- ASO (Antisense Oligonucleotide): A modern type of medicine designed to "silence" or turn off specific harmful genes.
- Sporadic ALS: The form of ALS that occurs randomly in the population without a clear family history, accounting for about 90% of all cases.
Jia, Q., Zhu, L., Li, D. et al. Caspase-4 transgenic mice exhibit cytoplasmic TDP-43 accumulation and age-dependent neuropathology. Nature Communications (2026).
DOI: 10.1038/s41467-026-73724-7 · 閱讀全文 →Read full text →
本頁為教育性整理,非原文翻譯;原文版權屬原出版方。An educational summary, not a translation; copyright remains with the original publisher.