一棵棵小樹依序修正前一輪的錯誤,再加權整合,輸出某人屬於 ALS 的機率。
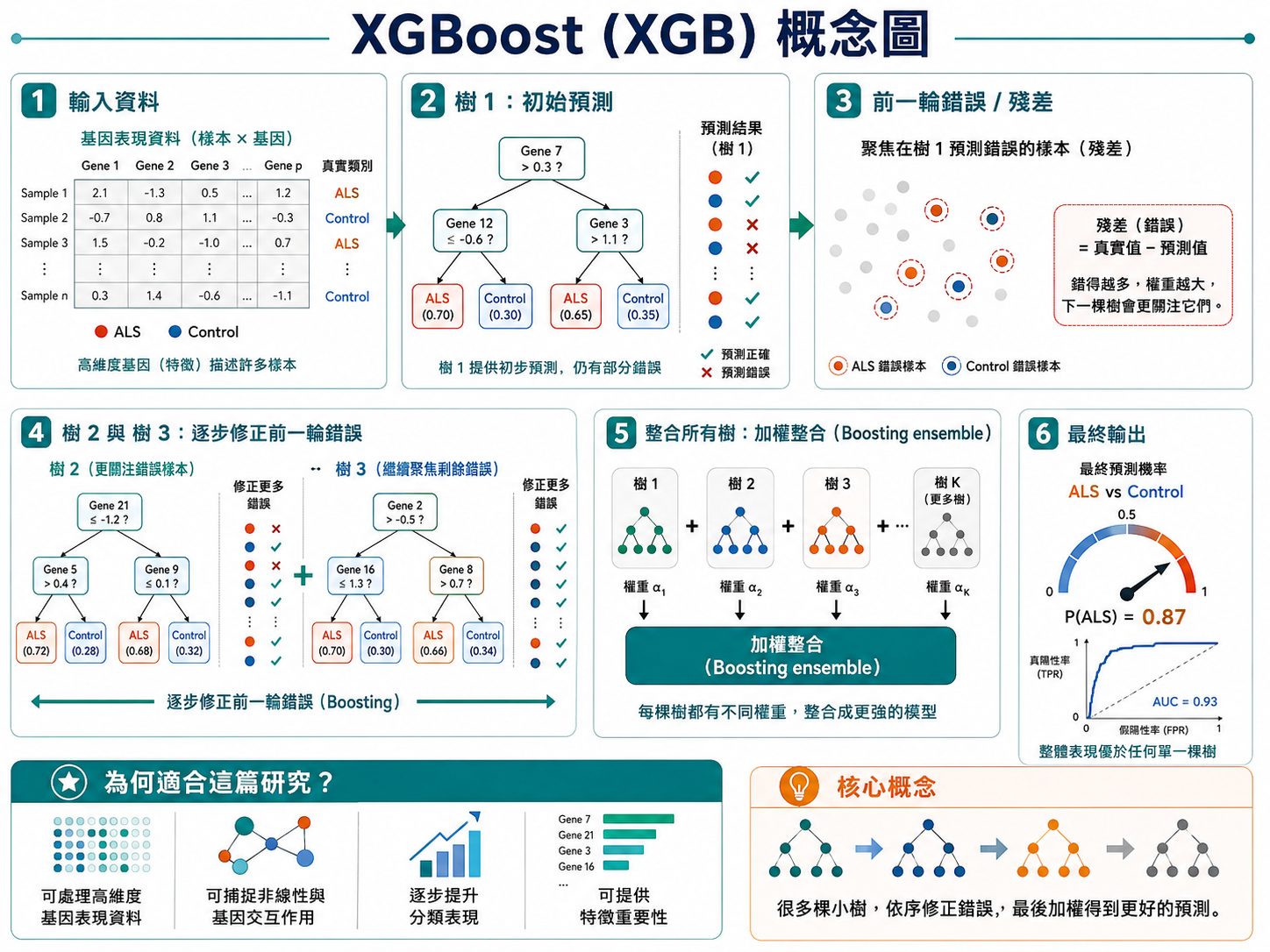
這張 XGBoost 圖想表達的核心問題很簡單:如果我們手上有很多 ALS 病人與健康對照者的血液 RNA-seq 資料,能不能只看這些基因表現,就判斷一個人比較像 ALS 還是 control?
這篇研究的做法,是先把每一位受試者的血液基因表現整理成一個大型矩陣。每一列代表一位樣本,每一欄代表一個基因,而每個格子中的數值,就是那個基因在該樣本中的表現量。對模型來說,這些基因就是「特徵」,而病人是否為 ALS,則是模型要學習的「答案」。研究者先從 RNA-seq 中找出 ALS 與 control 之間顯著不同的差異表現基因(DEGs),再把這些基因丟進機器學習模型中,看看能不能建立一個準確的分類器。文中比較了多種方法,最後發現 XGBoost 的表現最好,因此成為後續主要模型。
XGBoost 的運作方式,不是只用一棵決策樹做判斷,而是用很多棵小樹逐步接力。第一棵樹會先根據少數幾個基因,把樣本粗略分成 ALS 或 control。這時候它不可能完美,因此一定會有一些樣本被分錯。接下來,第二棵樹不會從頭亂猜,而是會特別去「注意前一棵樹容易分錯的樣本」。也就是說,模型會把上一輪的錯誤視為一種「殘差」或「尚未學好的部分」,然後讓下一棵樹專門去修正它。第三棵樹再繼續修正前面剩下的錯誤,如此一棵接著一棵,慢慢把整體預測變得更準確。這也是為什麼這個方法叫做 boosting:它不是單一模型一次做完,而是透過連續修正錯誤,逐步把模型「推強」。
所以,你在圖裡看到的「樹 1 → 樹 2 → 樹 3 → … → 樹 K」,其實不是很多獨立模型,而是一個會逐步學習、持續改進的模型系統。最後,這些樹的輸出會被加權整合,得到一個總分,並轉換成某個樣本屬於 ALS 的機率。假如最終輸出是 P(ALS)=0.87,意思就是模型判斷:這個人的血液基因表現型態,有很高機率屬於 ALS 那一群。
這篇論文之所以適合用 XGBoost,主要是因為 RNA-seq 資料很典型地具有高維度的特性:樣本數相對有限,但基因數很多。這種資料往往不是單一基因決定結果,而是很多基因之間共同作用,甚至存在非線性關係與交互作用。XGBoost 的樹模型很擅長捕捉這種複雜模式,例如某些基因必須同時高、某些基因必須低於某個閾值,分類訊號才會明顯。這種能力,比單純的線性模型更有機會抓到真實的疾病 signature。研究中作者先比較了 ridge、LASSO、elastic net、SCAD、MCP、L1/2 與 XGBoost,結果 XGBoost 在內部資料中的 AUC 最高,因此被選出來做後續優化。
接著,作者並不是直接把幾千個基因全部拿去做臨床模型,而是進一步做特徵篩選,建立較小的 gene panels。他們從不同 DEG 篩選條件中,最後得到 27-gene、30-gene、29-gene 三種 panel,並再組成 46-gene combined panel。這些 panel 在內部測試的表現都非常好,且在外部獨立 RNA-seq cohort 中,combined panel 仍然能得到不錯的 AUC,顯示這不是只在原始資料裡好看,而是具有一定的外部泛化能力。
換句話說,這張圖真正想告訴你的是:ALS 的血液轉錄體中,的確存在可以被機器學習擷取出來的分類訊號,而 XGBoost 是用來把這個訊號系統化、量化、並轉成可預測模型的工具。
如果再用一句更白話的話總結這張圖,可以這樣說:XGBoost 圖是在描述,研究者如何把「很多基因的複雜變化」變成一個能判斷 ALS 與 control 的預測器。
本頁為教育性整理,搭配主文圖解 XGBoost 的概念,非原文翻譯。原研究:Zhao, Y. et al. Gene expression signatures from whole blood predict amyotrophic lateral sclerosis case status and survival. Nature Communications 16, 9631 (2025). DOI